Creutzfeldt-Jakob disease: can an infectious protein be treated?
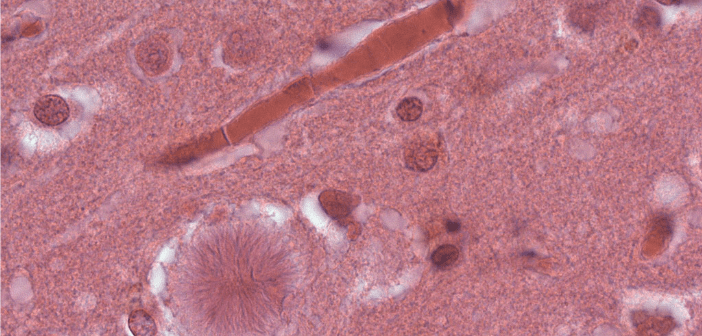
Creutzfeldt-Jakob disease (CJD) is a rare, degenerative and fatal brain disorder belonging to a family of human and animal diseases, known as transmissible spongiform encephalopathies, or prion diseases [1]. It was initially believed that a transmissible agent, such as a virus, was responsible for causing the disease. However, more recently the consensus has become that prions actually transform normal proteins into infectious molecules [2]. Although a transmissible agent is responsible for CJD, it isn’t considered to be contagious in a traditional sense. At present, four major categories of CJD have been identified (Table 1). The most common form of the...